Poster #P01
Adaptive Quantum Chemistry Basis Sets
Gaussian Type Orbitals (GTOs) from the Pople basis set family have been among the most widely employed basis functions in quantum chemistry calculations for five decades. We introduce a machine learning model that improves the STO-3G, 3-21G, 6-31G and 6-31G* basis sets by adapting basis functions to the local atomic environments prior to the start of Self Consistent Field (SCF) calculations. This is achieved by scaling the variance of the radial Gaussian functions leading to the atomic orbitals contracting or expanding based on the local chemical environment. Optimal scaling factors are calculated using the variational principle, i.e. by minimizing the Hartree-Fock (HF) energy, and form the training data for the machine learning (ML) model. After training on a rather small number of molecules, it is shown that using the basis sets predicted by the ML model provides more accurate atomization energies than default Pople basis sets in up to 99% of tested HF calculations.
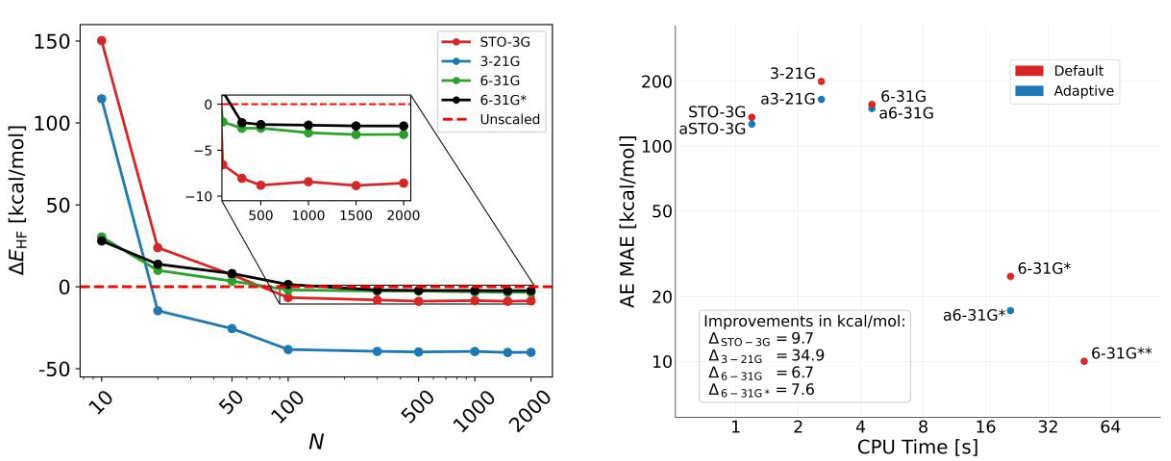
Figure 1. Left: Change in Hartree Fock total energy using basis sets with ML scaling factors as a function training molecules. Right: Atomization energy error vs. CPU time using default and adaptive Pople basis sets.
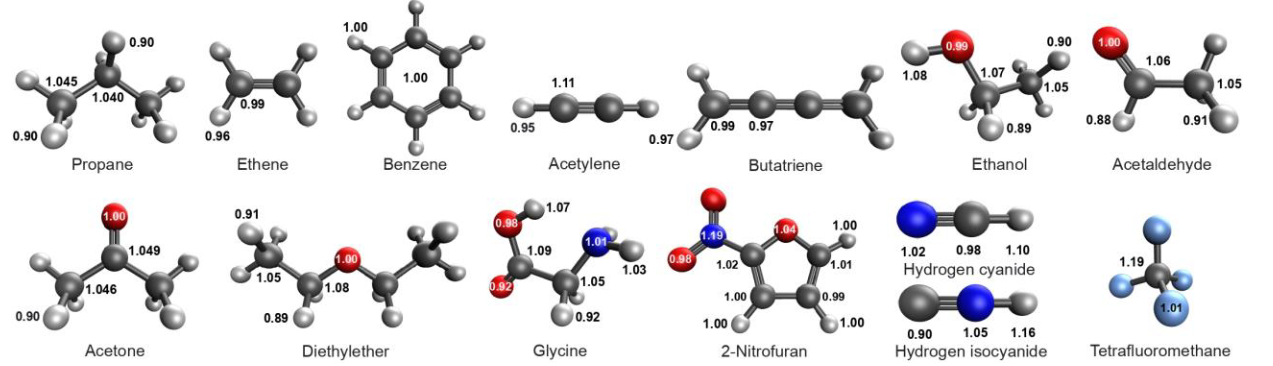
Figure 2. Molecules from the QM9 dataset with diverse functional groups and their optimal (HF energy-minimized) STO-3G valence orbital scaling factors. As can be seen the scaling factors are highly local and transferable and depend on the electron density near the atom.
Maximilian Ach
- HITS gGmbH
- Heidelberg University