Poster #P49
Reaction discovery with periodic boundary conditions using ab initio nanoreactor molecular dynamics
Selective catalytic reduction (SCR) is substantial for automotive and exhaust gas catalysis, reducing toxic nitrogen oxides to molecular nitrogen.[1] For the reduction of nitrogen oxides, many different possible formation mechanisms are supposed. However, during SCR dinitrogen oxide (N2O), which is a long-living greenhouse gas, appears as an unwanted side product.[2] The formation mechanism of N2O involves a chemical reaction network including complex reaction cascades. To discover unknown areas of this reaction network, methods of automated reaction discovery are essential. In this study, we employ the ab initio nanoreactor[3] method to investigate the various reactions occurring during the process of SCR to reveal a potential formation mechanism for dinitrogen oxide (N2O). To account for the periodic structure of the framework, ab initio nanoreactor molecular dynamics (AINRMD) calculations were extended by periodic boundary conditions (PBC) built in Turbomole.[4,5] Accelerated AINRMD simulations of a copper-exchanged zeolite with PBC were performed involving key reaction partners of the SCR, such as nitrogen oxides, ammonia and molecular oxygen. To increase reactivity, the gas phase molecules were accelerated towards the copper atom. The observed reactions are consistent with the literature and offer insights into a potential formation mechanism for N2O and a water-shuttled N2 formation. To elucidate the electronic structure, CASSCF calculations of key cycle members were performed.
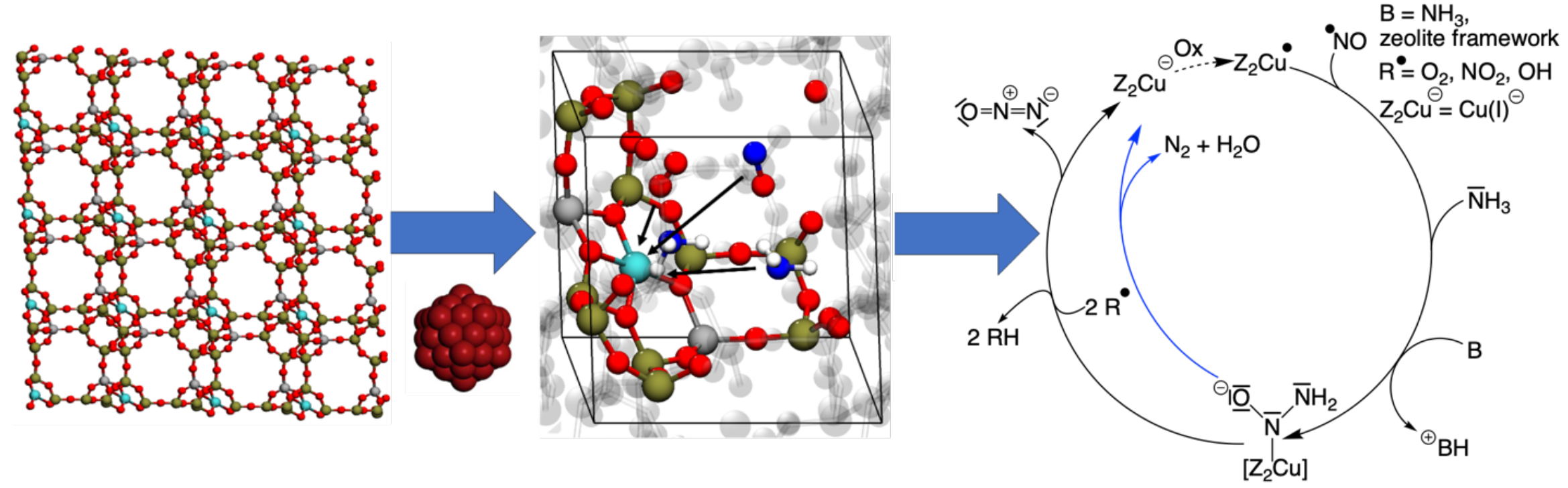
Figure 1. Left: zeolite crystal lattice structure. Middle: unit cell of the zeolite with periodic boundary conditions simulated using ab initio nanoreactor molecular dynamics. Right: discovered cycle for N2O (black) and N2 (blue) formation, with a dashed line indicating a reaction not yet studied.
- K. Khivantsev, J.-H. Kwak, N. R. Jaegers, et al., Chem. Sci. 2022, 13(35), 10383–10394.
- H. Ahari, M. Smith, M. Zammit, et al., SAE Int. J. Passeng. Cars - Mech. Syst. 2015, 8(2), 526- 530.
- L. P. Wang, A. Titov, R. McGibbon, R. et al., Nature Chem' 2014, 6, 1044–1048.
- R. Ahlrichs, M. Bär, M. Häser, et al., Chem. Phys. Lett. 1989, 162(3), 165–169.
- A. M. Łazarski, M. Burow, M. Sierka, J. Chem. Theory Comput. 2015, 11, 3029–3041.
Daniel Deißenbeck
- Institute for Physical Chemistry, Heinrich Heine University Düsseldorf, Germany