Poster #P35
Efficient interatomic descriptors for accurate machine learning force fields of extended molecules
Machine Learning Force Fields (MLFFs) enable modeling of chemical systems combining ab initio accuracy with the efficiency of mechanistic force fields. Despite the great success of ML methods, extending their applicability to larger molecules poses a challenge partly due to the rapid growth of the dimensionality of the descriptor. State- of-the-art descriptors include non-essential degrees of freedom or neglect long-range interactions by imposing a cut-off radius. Thus, finding a way to accurately describe both short- and long-range interactions without significantly increasing the descriptor size would advance ML modeling. In the present work, we propose a global descriptor optimization scheme that can be efficiently used to model large and flexible molecules. To achieve this, we developed a robust descriptor reduction methodology by employing sensitivity analysis. We analyse interaction patterns extracted from reduced GDML models of various systems of interest, including one supramolecular complex and units of four major types of biomolecules. Analysis of the relevant features demonstrates that some features included in local descriptors are not required for accurate models, while certain long- range features (even beyond 10 Å) can be crucial. These results pave the way to constructing global molecular MLFFs whose cost increases linearly, instead of quadratically, with system size.
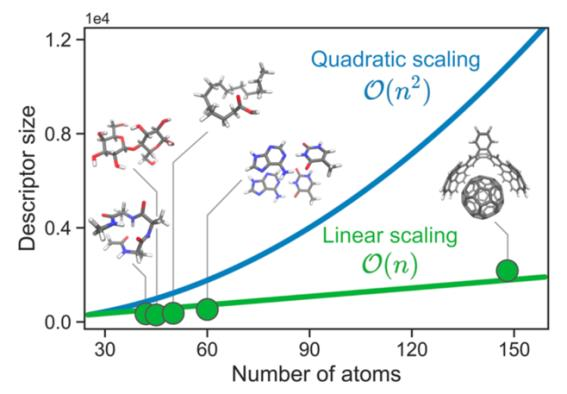
Figure 1. Scaling of the default and reduced global descriptors.
- Kabylda et al., Nat. Commun. 2023, 14, 3562.
- Chmiela et al., Sci. Adv. 2023, eadf0873.
Adil Kabylda
- University of Luxembourg