Poster #P42
The Bright Side of Machine Learning: from Ground- to Excited-State Applications
The performance of machine learning algorithms for electronically excited states significantly lags behind ground-state applications [1]. While the complexity of reference electronic-structure calculations poses a problem of its own, there are additional obstacles when learning both within the configuration space and across chemical compound space. In the configuration space, for example, we have to deal with insufficient smoothness of adiabatic states in the vicinity of conical intersections [2]. Moreover, most of the state-of-the-art molecular representations and machine learning models focus on extensive molecular properties such as the total energy, which is reflected in their design. These models often assume additivity of contributions from local atomic environments. However, this assumption does not hold for intensive properties such as excitation properties. On the other hand, global molecular representations encoding the molecule as a whole, usually struggle with missing permutational invariance and non-constant size when learning in the chemical compound space. We propose here a lightweight, yet expressive representation based on the expansion into spherical harmonics (see Fig. 1) and two machine learning kernels allowing its efficient application to both extensive and intensive properties. We demonstrate the performance of our kernel-based framework on multiple datasets, encompassing both chemical compound space and configuration space. Our approach is easily implementable thanks to its simplicity, and cheap to apply due to the small size of the representation and small number of hyperparameters. At the same time, it reaches or surpasses the accuracy of current state-of-the-art representations and kernels and stands out for its general applicability.
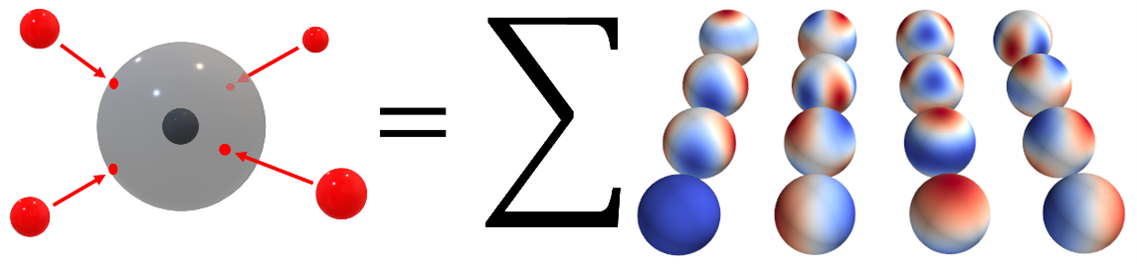
Figure 1. Schematic picture of the representation
- J. Westermayr, P. Marquetand, Chem. Rev. 2021, 121, 9873–9926.
- Š. Sršeň, O. A. von Lilienfeld, P. Slavíček, Phys. Chem. Chem. Phys. 2024, 26, 4306–4319.
Štěpán Sršeň
- University of Vienna