Poster #P24
Active Learning for Chemical Space Exploration
Machine learning models have been successfully developed for various applications, including different matters in chemistry [1]. While in many cases the performance of those models makes them more favorable over classical computational chemistry methods, their usage has a major constraint: the need for sufficient training data. This poses a serious limitation in contexts where labeled data is scarce or expensive to obtain. To overcome this issue, machine learning models can be trained with active participation. An active learning algorithm actively decides upon which training instance needs to be labeled next for the machine learning model to obtain the largest knowledge gain [2]. This process accelerates the convergence of model performance (Fig. 1) and therefore allows the usage of less training data, thus reducing labeling effort. A main performance factor of active learning methods is the criteria by which the model chooses the next training instance to be labeled. Numerous different selection strategies are known [3]. Selecting an active learning algorithm with superior convergence of model performance for a given dataset requires time for research, analysis and implementation. To bypass that obstacle we present an extensive benchmark of active learning algorithms.
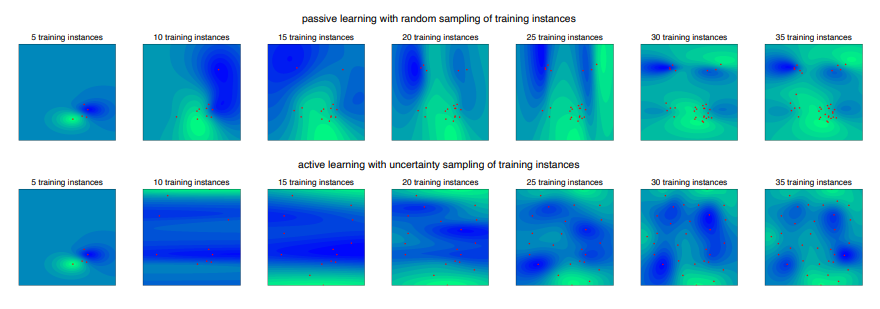
Figure 1. Performance comparison of active learning (uncertainty sampling method) and passive learning (random sampling method) on the Himmelblau function space.
- J. A. Keith, V. Vassilev-Galindo, B. Cheng, S. Chmiela, M. Gastegger, K.-R. Müller and A. Tkatchenko, Chemical Reviews 2021, 121, 9719-10240.
- B. Settles, Active Learning 2012. 1-20.
- P. Kumar and A. Gupta, Journal of Computer Science and Technology 2020, 35(4), 913-945.
Elizaveta Surzhikova
- Technische Universität Braunschweig